

我需要如何更新我的质量管理体系以符合FDA QMSR修正案?
通过马克Durivage,质量体系合规有限责任公司
 现在FDA已经正式宣布它的意图是协调和现代化医疗器械的21 CFR 820部分质量管理体系法规(QMSR),问题是:我需要对我的质量管理体系(QMS)做些什么?如果你的质量管理体系是基于ISO 13485:2016的要求,我建议下载21 CFR 820部分的质量管理体系,并分析你当前的质量管理体系可能需要更新的地方,以确保符合性。我建议你买一本ISO 13485:2016医疗器械-质量管理体系-监管目的的要求而且ISO 13485:2016 -医疗器械-实用指南,阅读它们,并进行全面的差距分析(假设你们当前的质量管理体系是基于21 CFR Part 820的要求)。
现在FDA已经正式宣布它的意图是协调和现代化医疗器械的21 CFR 820部分质量管理体系法规(QMSR),问题是:我需要对我的质量管理体系(QMS)做些什么?如果你的质量管理体系是基于ISO 13485:2016的要求,我建议下载21 CFR 820部分的质量管理体系,并分析你当前的质量管理体系可能需要更新的地方,以确保符合性。我建议你买一本ISO 13485:2016医疗器械-质量管理体系-监管目的的要求而且ISO 13485:2016 -医疗器械-实用指南,阅读它们,并进行全面的差距分析(假设你们当前的质量管理体系是基于21 CFR Part 820的要求)。
ISO 13485:2016包含八个主要条款,包括:
- 范围
- 引用标准
- 术语和定义
- 质量管理体系
- 管理责任
- 资源管理
- 产品实现
- 测量、分析和改进
每个条款都包含要求和子条款,子条款通过提供标准对质量管理体系要求的细节来支持主条款。就本文而言,21 CFR Part 820将被称为法规,ISO 13485:2016将被称为标准。我将强调标准各部分的差异,以及为符合更新后的法规而必须修改的程序,以及标准符合法规所需的额外要求。
1.范围
本标准适用于提供服务的组织,包括医疗设备的设计和开发、生产、存储和分销、安装或服务,以及提供产品的供应商,包括质量管理体系相关服务。该规定适用于成品设备和合同灭菌、安装、重新贴标签、再制造、重新包装或规格开发,以及执行这些功能的外国实体的初始分销商。生产医疗器械成品零部件的组织不要求采用该条例,但鼓励采用该条例。
FDA仍有能力批准豁免或差异;但是,这并不会减轻组织为了认证的目的而遵循标准的要求。
FDA仍将保持对组织的检查管辖权,无论其标准认证状态如何。此外,FDA不会对该标准或法规颁发认证。一般来说,如果一个组织是不受要求的21 CFR Part 807器械制造商和初始进口商的注册和器械清单在美国,无论ISO 13585认证状态如何,FDA检查您工厂的概率都可以忽略不计。
对要求范围的修改包括澄清,更具体的冲突法规仅在冲突的程度上优先。
此处所做的更改不应对质量管理体系文件产生任何影响,提供这些更改是为了澄清。
2.引用标准
ISO 9000:2015质量管理体系基础和词汇应在《质量手册》第2节《规范参考》中列出,以帮助审核员和检查人员进行审核和官方监管检查。
3.术语和定义
的术语和定义ISO 9000:2015质量管理体系基础和词汇,标准和法规将适用。但是,法规中的定义将优先。这些文件应列在《质量手册》第3节的条款和定义中,以及任何公司特定条款,以帮助审核员和检查员进行审核和官方监管检查。
4.质量管理体系
根据联邦食品、药品和化妆品法案第501(h)条,不符合法规中任何适用要求的设备被视为掺假。这种设备以及任何对未能遵守规定负责的人都将受到监管行动的约束。这是在ISO认证机构可能采取的任何行动之外。
该标准要求组织编制一份质量手册,这在以前的法规中并没有要求。质量手册应在较高的层次上描述组织如何遵守标准、法规和其他适用的法规要求。质量手册不应该是标准和规章的反刍。任何排除和不适用的应被记录,并提供适当的理由。最佳做法是包括一个合规矩阵,其中包含地图,显示您的QMS如何以及在何处满足标准和法规的每一项要求,以便在审核和官方监管检查期间帮助审核员和检查员。
法规要求制造商提供符合标准和法规要求的质量管理体系文件,并在适用时满足以下法规要求:
我建议在适用的情况下,在质量手册的规范性参考第2节中列出这些额外的法规要求。
你们的记录控制标准操作程序(SOP)必须要求批准或重新批准记录(纸质或电子)的每个人的签名和批准日期。
你们的记录控制SOP和/或外部检查SOP应规定被视为机密的记录应予以标记,以帮助FDA确定记录中包含的信息是否可以向公众披露。
5.管理责任
随着该标准的采用,将更加强调在整个产品生命周期中识别、分析、评估、控制和监测风险,以确保设备安全有效。管理层将负责确保在整个质量管理体系中考虑风险管理和基于风险的思想,包括计划、外包、设计和开发、可追溯性、采购控制、验收活动、生产和过程控制、服务、安装、数据分析以及纠正和预防措施(CAPA)。
该法规将最高管理者定义为有权建立或变更制造商质量方针和质量管理体系的制造商高级员工。这个职位以前被称为具有执行责任的管理人员。更新你的质量手册和标准操作程序,包括管理责任,管理评审,向监管机构报告等,以反映使用适当的术语,最高管理层。
6.资源管理
除了风险管理和基于风险的思维的潜在应用之外,标准的要求和法规之间不存在具体的差异。
7.产品实现
您的通信SOP应参考21 CFR Part 806 Medical Devices;纠正和移除的报告,以及在发生纠正或移除的情况下,贵组织将如何与客户和FDA沟通。
你们的设计和开发SOP将需要要求对II类、III类和某些I类设备应用设计控制。I类设备是指由计算机软件自动化的设备以及下表所列的设备:
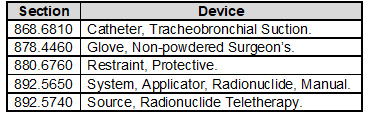
法规对设计评审要求对被评审的设计阶段不负有直接责任的个人的要求尚未转移到标准中。但是,您可能需要考虑将此最佳实践添加到设计和开发SOP中。
你们的生产控制和服务提供SOP将需要要求在设备历史记录(DHR)中记录每个医疗设备或批次医疗设备的唯一设备标识符(UDI)。
你们需要更新标签和包装SOP,以确保标签和包装在放行或储存前已经过准确检查,并在适用时包括以下内容:
- 正确的UDI或通用产品代码(UPC),或任何其他设备标识;
- 截止日期;
- 存储指令;
- 处理指令;而且
- 任何额外的处理指令。
使用标签的发布必须通过获得批准发布的每个人的签名(纸质或电子)和发布日期来记录。
你们的标签和包装SOP还必须确保已经建立和维护了标签和包装操作,以防止错误,包括但不限于,在使用前立即检查标签和包装,以确保所有器械都具有医疗器械文件中规定的正确标签和包装。标签检查的结果必须通过获得批准放行的每个人的签名(纸质或电子)和放行日期来记录。
您将需要更新您的维修SOP,以确保在维修活动中记录以下信息:
- 服务设备的名称;
- 任何UDI或UPC,以及任何其他设备标识;
- 送达日期;
- 维修设备的个人;
- 服务:提供的服务;而且
- 任何测试和检验数据。
你们的标识和可追溯性SOP将需要记录一个系统,根据21 CFR Part 830的要求为医疗器械分配唯一的器械标识。
您还需要更新您的识别和可追溯SOP。植入式医疗设备标准的可追溯性要求现在将额外适用于支持或维持生命的设备,或那些在按照标签中提供的使用说明正确使用时未能执行,可以合理预期会导致重大伤害的设备。此外,你们的识别和可追溯性SOP应参考21 CFR第821部分医疗器械跟踪要求,并定义适用的跟踪此类器械的流程。
8.测量、分析和改进
你们将需要更新你们的投诉处理SOP,以确保必须按照21 CFR第803部分医疗器械报告的要求向FDA报告的投诉,制造商确定必须进行调查的投诉,以及制造商调查的投诉,无论这些要求如何,至少捕获以下信息:
- 设备名称;
- 收到投诉的日期;
- 任何UDI或UPC,以及任何其他设备标识;
- 信访人的姓名、地址、联系电话;
- 投诉的性质及详情;
- 采取的任何纠正措施;而且
- 对投诉人的任何答复。
向监管机构报告SOP和/或针对交货后检测到的不合格产品采取的行动SOP应参考21 CFR第806部分医疗器械;纠正和移除的报告,以及在发生纠正或移除的情况下,组织将如何与客户和FDA沟通。
问题
如果您对从21 CFR Part 820到ISO 13485:2016的过渡有任何问题,请随时联系作者.
作者简介:
 Mark Allen Durivage曾担任执业医师、教育家、顾问和作家。他是Quality Systems Compliance LLC的管理首席顾问,ASQ研究员和SRE研究员。他在锡耶纳高地大学获得计算机辅助加工学士学位,在东密歇根大学获得质量管理硕士学位。他拥有多项认证,包括CRE, CQE, CQA, CSQP, CSSBB, RAC (Global)和CTBS。他写了几本书,可通过ASQ质量出版社,发表文章质量进步他经常为Life Science Connect撰稿。Durivage住在密歇根州的兰伯特维尔。请随意给他发邮件有任何问题或意见。
Mark Allen Durivage曾担任执业医师、教育家、顾问和作家。他是Quality Systems Compliance LLC的管理首席顾问,ASQ研究员和SRE研究员。他在锡耶纳高地大学获得计算机辅助加工学士学位,在东密歇根大学获得质量管理硕士学位。他拥有多项认证,包括CRE, CQE, CQA, CSQP, CSSBB, RAC (Global)和CTBS。他写了几本书,可通过ASQ质量出版社,发表文章质量进步他经常为Life Science Connect撰稿。Durivage住在密歇根州的兰伯特维尔。请随意给他发邮件有任何问题或意见。