

浏览FDA的紧急使用授权流程
通过比卡Chatterjee, Pharmatech Associates首席执行官
2020年1月31日,卫生与公众服务部部长(HHS)宣布全国进入紧急状态,以抗击艾滋病 由新型冠状病毒SARS-CoV-2引起的COVID-19大流行。因此,FDA被授权根据其2017年指南中定义的标准授予紧急使用授权(EUAs)1适用于任何有助于SARS-CoV-2诊断、预防或治疗的医学对策。这将影响内部生产和药物赞助商使用的CDMOs。以下是影响和细节的详细介绍。
由新型冠状病毒SARS-CoV-2引起的COVID-19大流行。因此,FDA被授权根据其2017年指南中定义的标准授予紧急使用授权(EUAs)1适用于任何有助于SARS-CoV-2诊断、预防或治疗的医学对策。这将影响内部生产和药物赞助商使用的CDMOs。以下是影响和细节的详细介绍。
欧洲大学协会是什么?
FDA将EUA定义为“在涉及化学、生物、放射性或核(CBRN)制剂的紧急情况下,对未经批准产品的加速授权和使用,或对已批准产品的超说明书使用”。2这些mcm可以包括药物、生物产品和设备,这些药物、生物产品和设备有可能“在没有足够的、批准的和可用的替代药物时,诊断、治疗或预防由CBRN制剂引起的严重或危及生命的疾病或状况”。
FDA指南定义了EUA流程的六个要素,如下图1所示:
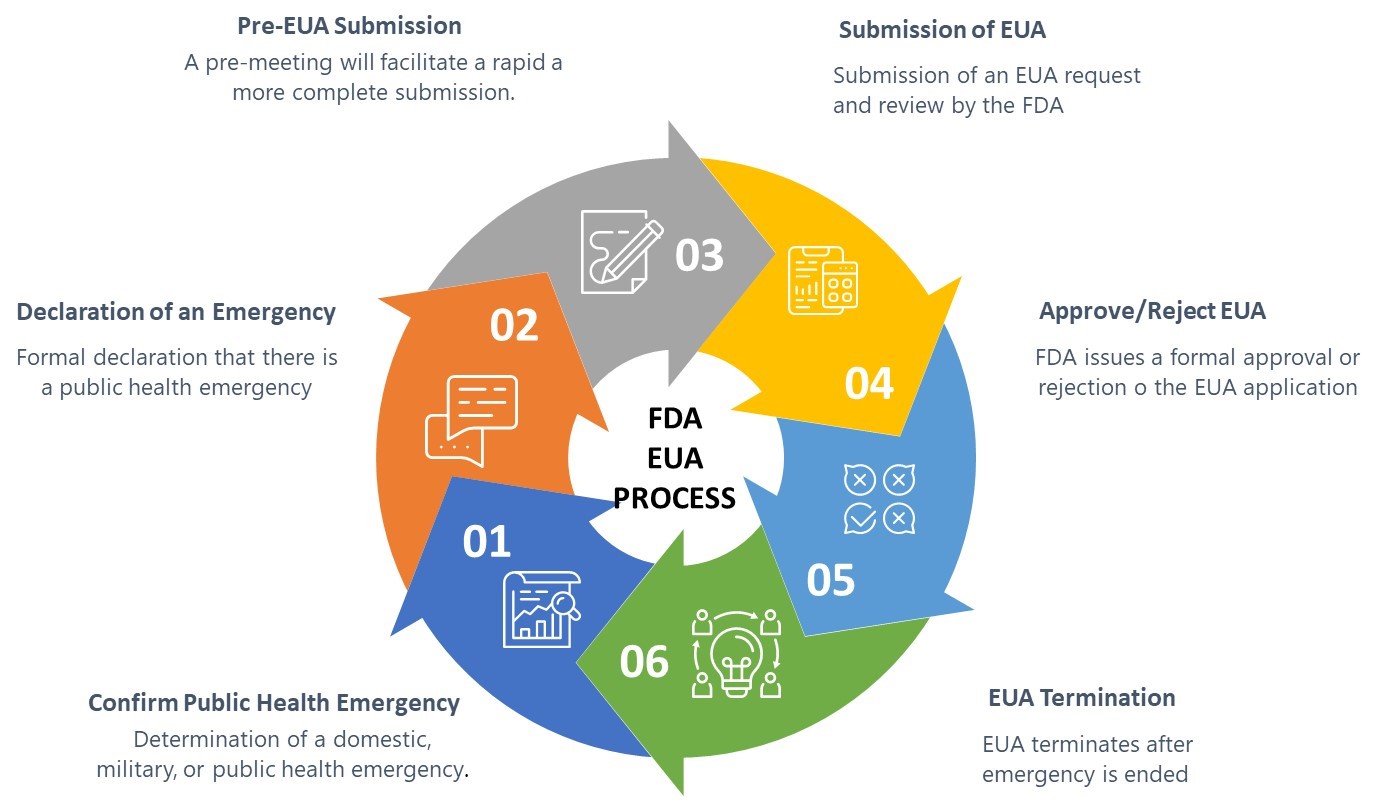
图1:FDA紧急使用授权程序
符合EUA标准
FDA制定了参与EUA计划和获得EUA必须满足的四个宽泛标准:
- 出现严重的或危及生命的情况
- 有效性的证据
- 风险-效益分析(安全)
- 没有其他办法来解决这种危及生命的状况
存在危及生命的情况
具体来说,为了让FDA发布EUA,美国卫生与公众服务部部长的EUA声明中提到的CBRN制剂必须能够引起严重或危及生命的疾病或状况。
有效性的证据
对于EUA来说,这一要求比正式法规提交中通常适用的标准要低,特别是只要求拟议的MCM“可能有效”地预防、诊断、或治疗严重或危及生命的疾病或状况,这些疾病或状况可能由美国卫生与公众服务部部长根据联邦食品、药品和化妆品法案(FD&C)第564(b)条宣布的紧急情况或紧急威胁中确定的CBRN制剂引起。FDA打算通过风险-收益分析逐个案例评估其潜在有效性。如果,基于现有的全部科学证据,有理由相信该产品可能对指定用途有效,FDA可以批准其紧急使用,前提是还满足签发EUA的其他法定标准。
风险-效益分析
在确定该产品的已知和潜在好处是否大于风险时,FDA将查看所有可用的科学证据,以做出总体风险-收益的决定。证据可能来自多种可供FDA考虑的来源,如国内外临床试验的结果、动物模型的体内疗效数据和体外数据。FDA建议EUA申请包括对候选产品已知的和潜在的风险和益处的讨论,其中包括对上述要求的数据和信息的综合,包括:
- 为降低风险或优化效益而采取的措施
- 局限性、不确定性和数据缺口
- 描述不应使用本产品的情况(如禁忌症)
- 在已知的范围内,涉及CBRN代理(实际或潜在)所构成的威胁的信息,以及与风险和收益评估相关的任何预期响应和操作考虑。
虽然FDA有意提议放宽cgmp下产品开发和生产的典型质量框架,但它仍然需要验证有效性和安全性。例如,通风机制造商仍需证明其符合适用的共识标准。然而,符合性演示的水平可以公开讨论,这是与FDA进行eua前提交会议非常有帮助的一个例子。事实上,FDA强烈建议尽早与该机构接触任何潜在的EUA产品,以促进更完整的EUA申请,并增强FDA审查和最终批准EUA的能力。
虽然在国家紧急情况下时间是至关重要的,但快速前进并增加患者的风险是FDA最关心的问题。一般来说,FDA建议提交eua前活动的数据应遵循提交IND前、IND和设备预提交的建议。根据我们的经验,这对于确保FDA能够放心地使用风险缓解数据是非常宝贵的,特别是当提交支持通过非传统途径开发的mcm的数据时,如学术界或生命科学行业以外的制造商。具体来说,传统的控制,如要求处方豁免大规模分发治疗或要求正式的风险评估和缓解策略(REMS)计划,经常在这些早期会议中讨论和最初论证。
没有其他的选择
对于FDA签发EUA,必须没有足够的、已批准的和可用的替代候选产品用于诊断、预防或治疗疾病或状况。如果供应不足,无法完全满足紧急需要,则可能认为一种潜在的替代产品“不可用”。潜在替代产品可能被视为“不足”,例如,有特殊情况或禁忌数据人群(如儿童、免疫力差的人,或者个人药物过敏),如果一个批准的剂型产品不适合使用在一个特殊的人口(例如,一种针对不能吞咽药片的人的药片),或者是否该制剂对已批准的和可用的替代产品有或可能有耐药性。
欧洲大学协会提交
EUA模板可从FDA网站下载。FDA建议EUA申请应包括一份关于该产品安全性和有效性、风险(包括不良事件概况)和益处的现有科学证据的组织良好的摘要,以及任何可用的、已批准的该产品替代品。支持EUA所需数据的确切类型和数量可能因已宣布的紧急情况或紧急威胁的性质以及候选产品的性质而异。FDA可能会根据具体情况寻求额外的数据和信息,以确保符合签发EUA的法定标准。具体来说,EUA提交将要求:
- 产品和预期用途的说明。这应包括识别产品可能有效的严重或危及生命的疾病或状况;预期在何处、何时、如何使用产品;和/或产品可能用于的人群。
- 产品的FDA批准状态的描述,其中应包括产品或预期用途是否处于研究申请中。
- 未满足需求的总结,包括任何已批准的替代产品的识别及其拟议用途的可用性和充分性,以及EUA将解决的未满足需求。
- 有效性和安全性信息,包括来自任何临床研究的数据和任何不良事件监测要求的非临床体内和体外数据。
- 风险和收益的分析,包括为降低风险或优化收益所采取的任何措施、任何禁忌症和数据中的任何缺口。(该分析应通过一个结构化的风险分析过程或框架来导出,该过程或框架设计为MCM的预期用途提供适当的解决方案。)
- 关于你们产品的化学、制造和控制,以及任何可用的和批准的产品替代品的信息。
- 提供有关给药剂量、禁忌症、警告和不良事件的信息的情况说明书,以便分发给医疗保健人员和您的产品的授权配药商。
审查和批准的时间将根据提交的需求在很大程度上的紧迫性以及为减轻潜在风险而提交的EUA的完整性而有所不同。正如指南中所述,FDA还为其审查EUA请求确立了优先级。
EUAs紧急失效
重要的是要认识到,在EUA下批准的MCM并不意味着FDA批准、许可或许可。由于HHS EUA声明的终止而导致EUA终止时,未经批准的产品或其标签,以及未经批准使用已批准产品的产品信息,必须按照fda和c法案第564(b)(2)(b)(3)59条处理。FDA鼓励赞助商继续开发他们的产品,努力在紧急情况结束后获得FDA的批准。
结论
通过EUA程序的规则与成功提交监管文件的原则没有太大不同。尽早与FDA建立强有力和清晰的对话将确保申诉人能够解决MCM预期使用的主要风险。认识到EUA不是免费通行证是很重要的,特别是对于通过非传统方法开发的mcm。了解FDA的期望将允许赞助商充分利用在EUA期间获得的可用数据,并可能在EUA被撤销后使其处于更顺畅的监管流程中。对于想要利用合同服务提供商的MCM开发者来说,在质量协议和供应协议中明确知识产权等关键问题是极其重要的。这一点对于刚进入市场的开发者和非在传统cGMP框架下生产的产品的责任尤其重要。
引用:
- 医疗产品和相关当局的紧急使用授权:行业和其他利益攸关方指南(反恐和新出现威胁办公室)https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization
- https://www.fda.gov/medical-devices/emergency-situations-medical-devices/emergency-use-authorizations
关于作者:
 Bikash Chatterjee是Pharmatech Associates公司的首席运营和科学官。他在制药、生物技术、医疗设备和IVD产品的设计和开发方面拥有超过30年的经验。他的工作指导了美国和欧洲十几种新产品的成功批准和商业化。查特吉是USP国家顾问委员会的成员,也是美国质量协会金门分会的前任主席。他是《在制药工业中应用精益六西格玛》一书的作者,也是国际会议的主讲人。Chatterjee持有the University of California at San Diego的生物化学学士学位和化学工程学士学位。
Bikash Chatterjee是Pharmatech Associates公司的首席运营和科学官。他在制药、生物技术、医疗设备和IVD产品的设计和开发方面拥有超过30年的经验。他的工作指导了美国和欧洲十几种新产品的成功批准和商业化。查特吉是USP国家顾问委员会的成员,也是美国质量协会金门分会的前任主席。他是《在制药工业中应用精益六西格玛》一书的作者,也是国际会议的主讲人。Chatterjee持有the University of California at San Diego的生物化学学士学位和化学工程学士学位。